
FDA Software Validation: What You Need To Do To Validate Your Quality Computer Systems

By Penny Goss, Technical Solutions
The FDA (Food and Drug Administration) and IEC (International Electrotechnical Commission) requirements for validation of your manufacturing and quality system software can conjure up a lot of questions. Understanding the actual guidelines and best practices for meeting these requirements isn’t always clear and, as a result, your software may be compliant but you may not be.
This article provides answers to the top five most common software validation and documentation questions asked by others in FDA-regulated industries and demonstrates best practices for meeting the guidelines.
What Systems Are Considered Quality Systems?
The FDA mandates software used for the design, manufacture, packaging, labeling, storage, installation, and servicing of all finished devices intended for human use shall be validated. While ISO (International Organization for Standardization) and SOX (Sarbanes-Oxley) regulations are not as clear about the validation process, they do require software development lifecycle processes be followed in areas where they apply because software validation demonstrates evidence which provides a high degree of assurance that a specific process will consistently produce the expected result.
Your initial system assessment should provide you with a list of systems which require validation. Below are eight of the most common systems, all of which fall under FDA regulation as well as ISO and SOX control. The systems in red typically affect multiple business units within the organization, most of which are Configurable-off -the-Shelf (COTS) software systems. These systems allow you to configure the software to meet your business needs.
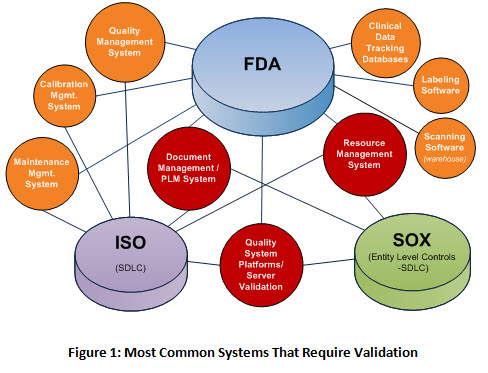
What Documentation Is Required For Regulatory Validation?
The following documents are what auditors like to see in a Quality System Validation:
Software Validation Protocol; Network Diagram; Software Requirements Specification; Risk Analysis (the GAMP standard template is recommended); Part 11 Compliance Analysis; Design Specification (only for systems or areas of the system which contain custom code such as integrations between your Product Lifecycle Management (PLM) and Enterprise Resource Planning (ERP) systems) ; Test Plan; Test Specifications/Test Cases; and a Final Validation Report. These documents may be combined so long as you capture all of the information.
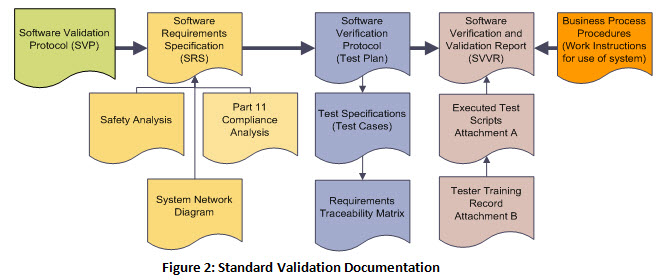
- Software Validation Protocol (Validation Plan)
This document outlines the project deliverables and responsibilities. - System /Software Requirements Specification
This document details system requirements. However, this is more than just a list of functional requirements it also should capture a good description of the various components that make up the system so that everyone has a clear understanding of what this system involves. The Requirements Specification also needs to include information around physical hardware requirements, physical software requirements, client user requirements, training requirements, and detail about any customizations or integration with other systems. - Network Diagram
This required document provides a visual layout of how the system is configured on the network. It serves to demonstrate that you understand how your system is configured for your implementation. (Note: this diagram may be included as an attachment to the Software Requirements Specification.) - Risk Analysis
This document evaluates application safety and identifies potential hazards, the causes and the effect each hazard has on the application safety and use. Because this is a business system, the risk assessment should focus on the business processes being managed by the system versuss a more traditional FMEA risk assessment for software programs which are part of a device and pose direct patient risk. Not all risks will be solely mitigated by the software, some risks are mitigated procedurally. - 21 CFR Part 11 Compliance Analysis
This document evaluates system applicability/requirements for the use of electronic signature as required by the FDA in 21 CFR, Part 11. - Design Specification
Typically a design specification is not required for a purchased configurable business quality system. In the event that major integration or customization is to be performed as part of the project this document may be added as deemed appropriate by the project team and quality reviewer. - Verification Protocol (Test Plan)
This document defines the type of testing to be completed along with the procedures and schedules for those tests. - Test Specifications (Test Cases)
This document contains the system level test cases, based on the functional requirements set forth in the Requirements Specification. If these are separate or maintained as an attachment to the Verification Protocol it makes it easier to add modules or new phases to the validation package while limiting revision time. Plus they are easier to format and work with testing wise. - Requirements Traceability Matrix
This matrix details all system requirements including Requirements ID, and links them to the Test Case IDs. This is a required document that can be helpful going forward when a change occurs, as it makes it easier to assess and identify where there is impact. - Final Validation Report
The validation report should provide a summary of all documentation associated with the validation of the software and test case results. This report should include both a summary of all the validation activities and define how the system will be managed in production. Information such as what work instructions are used to train users to use the system, what system support is available, how the system will be backed up, and how change control will be managed are extremely important elements captured in this document. In essence it puts a bow on the validation package.
Why Can’t I Just Purchase Validation Documentation?
The FDA requires verification and validation activities cover how a system is configured in the customer’s environment so there is no one-size-fits-all validation. Many times software vendors will try to sell prepackaged validation documentation. Because a vendor selling a prepackaged validation does not know your requirements they try and list every functionality that the system has and let you whittle down the list of requirements based on your use of the system. Since they do not know exactly which requirements you are going to keep and which requirements you are going to throw out they can’t group the test cases to contain multiple requirements. Again, it looks like a great deal because they can show you all of these test cases that you are going to get with their packaged validation. What they aren’t telling you is that sorting out which ones you want and don’t want takes a lot of time (possibly weeks) that your team might not have.
Everyone’s business is a little different so software manufacturers such as Omnify Software have developed highly configurable systems that can be set up to manage your business processes electronically. Even if you don’t need to validate your system from a regulatory standpoint, methodical documentation and testing of your configuration is a good business function. It’s risky not to be sure (with documentation) that important activities are being handled electronically the way you think they are.
How Do I Upgrade Or Add Modules To A Validated System
If you designed your validation documentation well, it should be a fairly straight forward change control process to upgrade to new software versions or add new modules/functionality to your system. If it has been less than one year since the original validation, you should be able to revise the documents by adding requirements for the new functionality into the Software Requirements Specification under the functional requirements section and updating other validation documents accordingly. Then either append the original test case document or add new module test cases as an attachment. Keep in mind you should always retest some of the security elements to make sure there was no impact as well as verify that history files are maintained for new functionality that has a quality impact.
If it has been more than one year since the original validation, follow your change control procedure and include an assessment of other changes which may warrant full revalidation. Typically the software manufacturer has a major new software release that you will want to include or you may need/want to upgrade your infrastructure. In this case you need to review the previous validation, add new functionality, update the test cases to match current business processes, or update work instructions and re-test. Again, this is when those process-based test cases pay off.
Conclusion
The FDA requires that software systems used for quality purposes in place of paper records be validated for their intended use [Title 21 CFR Part 820(i)]. This means that when using COTS systems, companies must verify that the software is configured correctly to meet their business needs. Even if you are intending to do a relatively vanilla implementation you need to have documented evidence verifying that the system was correctly installed and configured correctly for your intended use. Performing software validation right the first time will save medical manufacturers both time and money now and in the future.
About The Author
For the last 15 years, Penny Goss has worked as a consultant and focused her business on verification and validation of technology controls and procedures to ensure compliance for businesses operating in regulated environments, primarily medical device, pharmaceutical, and biotech companies regulated by the FDA, ISO, and Sarbanes Oxley. Penny has helped numerous Omnify Software customers with their validation procedures with outstanding audit results.